My Ruby pipelines: Variant call (2)
- Agathe
- Jan 26, 2018
- 4 min read
Updated: Feb 5, 2019
Once reads are aligned to a reference genome or reference genes, the identification of SNPs (Single Nucleotide Polymorphisms) is possible. The aim then is to explore variations between individuals.
Welcome to the second post from the "Ruby pipelines" series.

(Artwork: Variations in Gems - Steven Universe)
Software used
samtools-1.3.1
vcftools-0.1.14
Linux commands
grep
sed
Ruby and Rake syntax
ENV[]
file
FileList
join
pathmap
puts
task
Main aims
The overall aim of the script described here, is to get a multisample VCF file ready to be fed to SHAPEIT version 2. For this, we need to:
Create a list of loci or chromosomes to be analysed
Call variants from all samples and get a multisample VCF file
Filter the VCF file (based on DP [read depth], removing INDELS and sites with missing data)
Next, SHAPEIT will be used to phase variants before the reconstruction of haplotypes.
Link to script: 2_variant_call.rake
Beginning of the script

Arguments
We provide three arguments to the script "2_variant_call.rake", using ENV:
fasta: the reference FASTA file
vcf_basename: the name you want for your multisample VCF file, without an extension
loci: as before, the type of samples you want analysed
In addition, as in script "1_mapping_to_targets", we give the list of FASTQ (read) files with FileList and we prepare the names and locations for our raw and filtered BAM files with pathmap.
Usage
To call the script, I would use a command like the one below. Of course you would need to adapt the name and location of the script and the FASTA file as necessary, and you may use whatever value you like for the vcf_basename and loci arguments. One thing you need though is to use the same arguments throughout scripts 1-4 from the "My Ruby pipelines" series.
rake -f scripts/2_variant_call.rake fasta=input.fas vcf_basename=multisample_vcf loci=domestication --trace
Task 1: Loci list
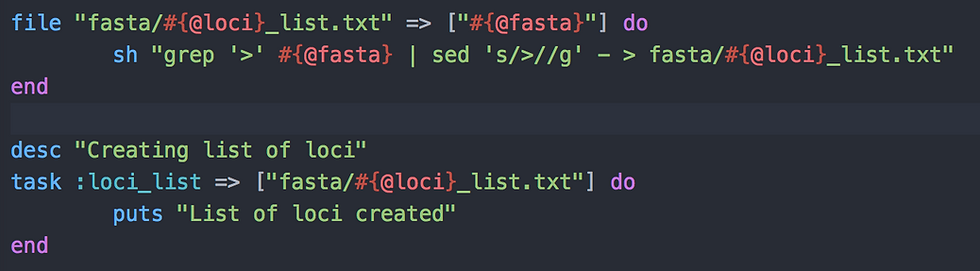
The first task is really simple and only need two basic Linux commands: grep and sed. What we do is:
With grep, select all lines with a "greater than" symbol; that is all lines with the loci or chromosomes name in a FASTA file
Remove that "greater than" symbol with sed to only keep the loci or chromosomes name
Print the resulting names in a file called domestication_list.txt (#{@loci}=domestication, see usage above) and stored in the folder "fasta"
The task is called :loci_list and requires that file described above, to be completed. It has this simple architecture I was talking about in the first post of the "My Ruby pipelines" series:
file "outfile" => ["required_input"] do
sh "command"
end
desc "description of my task"
task :my_task => ["outfile"] do
puts "my task is done"
end
Task 2: Calling variants
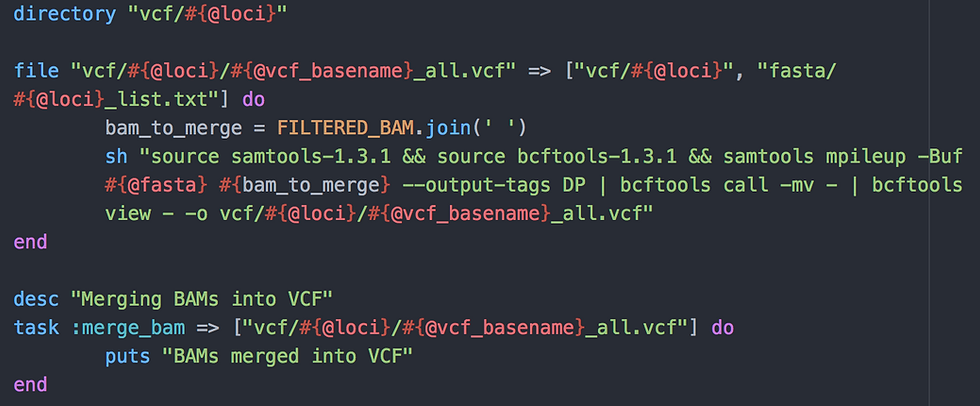
Here, we use a file task to create the file "multisample_vcf_all.vcf" (#{@vcf_basename} = multisample_vcf, see usage above) in a newly created folder called "vcf/domestication". The task requires that folder + the list of loci from the previous step.
I then use the mpileup command from SAMtools version 1.3.1 to call sequenced bases from all samples. This command needs the BAM files created with the previous script (1_mapping_to_targets.rake) but because I do not want to have to type the name of all BAM files manually, I need to initiate a new variable "bam_to_merge" in which I store these, that is all elements of my predefined FILTERED_BAM array, joined by a space. Rake would read something like:
samtools mpileup -Buf fasta/input.fas bam/filtered/bam1_best_lte0.25-clipped.bam bam/filtered/bam2_best_lte0.25-clipped.bam bam/filtered/bam3_best_lte0.25-clipped.bam bam/filtered/bam4_best_lte0.25-clipped.bam [...] --output-tags DP ...
For the mpileup command, I use the flags (from the SAMtools manual):
B: Disable probabilistic realignment for the computation of base alignment quality (BAQ). BAQ is the Phred-scaled probability of a read base being misaligned. Applying this option greatly helps to reduce false SNPs caused by misalignments.
u: Generate uncompressed VCF/BCF output, which is preferred for piping.
f: The faidx-indexed reference file in the FASTA format. The file can be optionally compressed by bgzip.
I also use the --output-tags DP option so that SAMtools output the DP tag, which is the read depth at base position, necessary to filter out variants with too low a read depth. I finally use the call and view commands from BCFtools version 1.3.1 to output variants only and convert the resulting BCF file to a VCF, respectively.
For the call command of BCFtools, the parameters are as follows (from the BCFtools manual):
m: alternative model for multiallelic and rare-variant calling designed to overcome known limitations in -c calling model (-c calling model is the previous variant calling model from BCFtools, pre-2016)
v: output variant sites only
The task is called :merge_bam and requires the multisample VCF file.
Tasks 3-5: VCF file filtering
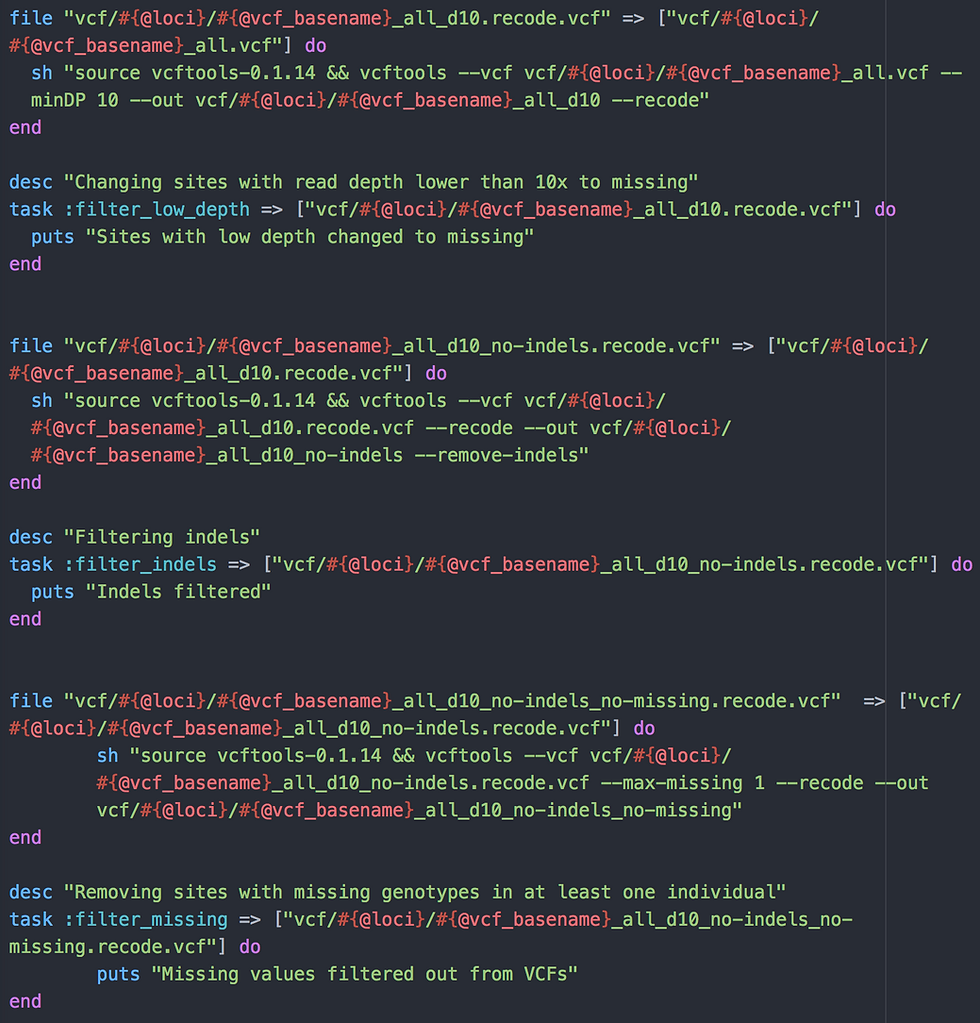
Three file tasks here to filter variants from our multisample VCF file using VCFtools version 0.1.14. Variants are removed if:
Read depth (DP) is greater than 10x - option --minDP 10
Variants are indels (insertions or deletions; SHAPEIT which we are going to use net does not like these) - option --remove-indels
One individual or more has missing data at the position (SHAPEIT does not like it either) - option --max-missing 1
End of the script

The default task requires the last filtering task described above. If all goes well, Rake will read the script up to the beginning and start from the first task to this last one.
Using this multisample VCF file, you can do a lot of things, such as:
Build a phylogenetic tree
Run a Principal Component Analysis (PCA) or Admixture (package in R) to study population structure
Compute sequencing statistics
Annotate variants to predict effect on transcripts
However, in this series of scripts, I am interested in reconstructing haplotypes from all individuals to then investigate genetic diversity between and within groups of these individuals. For this, I need to first phase the variants stored in my multisample VCF file and then to compute various statistics using DNAsp and including heterozygosity, genetic diversity Pi, Tajima's D or dN:dS.
In the next post of the "My Ruby pipelines" series, I will describe how to phase variants from a VCF file using SHAPEIT2.


Comments