My Ruby pipelines: Mapping reads to reference sequences (1)
- Agathe
- Jan 26, 2018
- 6 min read
Updated: Feb 5, 2019
This is the first post of the "Ruby pipelines" series in which I explain how I deal with basic NGS data analyses using the Rake build language.

Aligning reads to a reference genome or to a set of reference genes is one of the first step scientists need to achieve when dealing with NGS data. It aims at putting all the bits and pieces you have sequenced to their rightful places, similar to building a puzzle. You usually do this for multiple read sets or individuals, so that you can compare them to each other, identify variants that are shared or rare, build phylogenies, test for selection...
Many software are available for mapping
and manuals need to be read carefully to avoid bad surprises. After many trials and errors, I designed a pipeline that I am happy to work with and which I am going to share with you. However, it is important that everyone check their alignments at
different stages of the pipeline to potentially adjust the parameters used. I will first list the main aims of the pipeline as well as the software used to then explain my script step by step.
(Art by Mihai Criste: "Rien ne se perd... tout se transforme!")
Software used
bwa-0.7.4
samtools-1.3.1
ngsutils-0.5.7
Ruby and Rake syntax
each
ENV[]
file
FileList
multitask
pathmap
puts
task
transpose
zip
Main aims
Indexing your reference genome or sequences which should be in fasta format
Mapping your reads (in this example, they are 150 bp paired-end reads from HiSeq) to your indexed reference to get BAM files
Filtering the BAM files
Pretty simple, right?
Link to script: 1_mapping_to_targets.rake
Beginning of the script
I first need to give my script anything that is necessary for the analysis. This means writing the location of my read files (in FASTQ format, can be gzipped) and the location of my reference FASTA file. I also set my script as a rakefile, write my name and the date along with the usage for the script.
This is done so:

Usage
To start the script, you need to refer to the usage provided as a comment, line 3. In this example, I first source rake version 2.3.1 (not always necessary). Then, I call rake with my script using the "f" flag and I give the path to my reference FASTA file. I also add a variable called "loci" which is the type of genes I am analysing. I find this allows a better organisation of the data, as I need this analysis repeated for different types of genes.
In the end, I would run my script using a command similar to this:
source rake-2.3.1; rake -f scripts/1_mapping_to_targets.rake fasta=fasta/input.fas loci=domestication -j 70 ---trace
*domestication designates genes that were potential targets of selection during the domestication of plants. This is just an example and you may change this value to anything that you want.
*the "j" flag enables multiple sets of read files to be analysed in parallel. In this example, I am analysing 70 pairs of read files.
*trace, with two dashes before, will turn on the verbose mode of rake. I always set it on because it helps debugging the script would something go wrong.
*It is possible to add the name of a task if you do not want to go over all of the tasks described in your script. If so, you would need to add, just after "rake", a semicolon followed by the name of the task - no square brackets.
Input files
In rake, you have several options to load input files, including:
ENV["fasta"] ? @fasta = ENV["fasta"] : nil will require a variable called fasta which could be a file (path required) or just a name (see domestication above)
READ1 = FileList['reads/*_1.fq.gz'] will create an array of all files matching given conditions; here they need to be in a folder called "reads" and end with "_1.fq.gz". These are gzipped FASTQ files.
Output files
Because I am analysing many files at once, I cannot provide hard-coded names for my output files. They need to stem from their corresponding input files. For this, I use the pathmap method which will "map the path according to the given specification" (see pathmap method in the Ruby docs).
BAM = READ1.pathmap("bam/raw/#{@loci}/%n") will create a new array called BAM with files which names derive from those stored in READ1. These are BAM (alignment) files which are stored in "bam/raw/domestication/" (#{@loci}=domestication, see usage). They will have the same name as my FASTQ files, without the extension (=%n, see the Ruby docs above for more details).
Using the same method, I also provide names for my filtered BAM files. These will be stored in "bam/filtered/domestication" (see the raw to filtered substitution below) and they will have their own extension "_best_lte0.25-clipped.bam".
FILTERED_BAM = BAM.pathmap("%{raw,filtered}p_best_lte0.25-clipped.bam")
Task 1: Indexing FASTA files
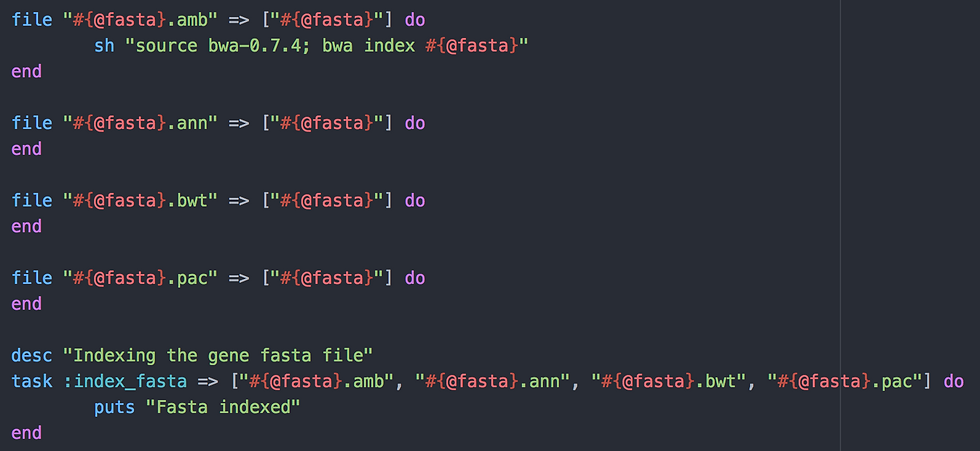
Here I use 4 file tasks to create four indices from my reference FASTA file:
fasta/input.fas.amb
fasta/input.fas.ann
fasta/input.fas.bwt
fasta/input.fas.pac
This is done using one command bwa index #{@fasta} which rake will translate to bwa index fasta/input.fas.
All file tasks require the original FASTA file, of course. This is coded by "=> ["#{@fasta}"]". If the FASTA file does not exist, the script will fail. To trigger the bwa command, I created a task called "index_fasta" that require all four indices. Rake will try to find out how to create the files, will see the file tasks and then the bwa command.
If all goes okay, rake will output "Fasta indexed".
Ultimately, the architecture I use for all my rake tasks is, more or less:
file "outfile" => ["required_input"] do
sh "command"
end
desc "description of my task"
task :my_task => ["outfile"] do
puts "my task is done"
end
Task 2: Mapping reads to a reference
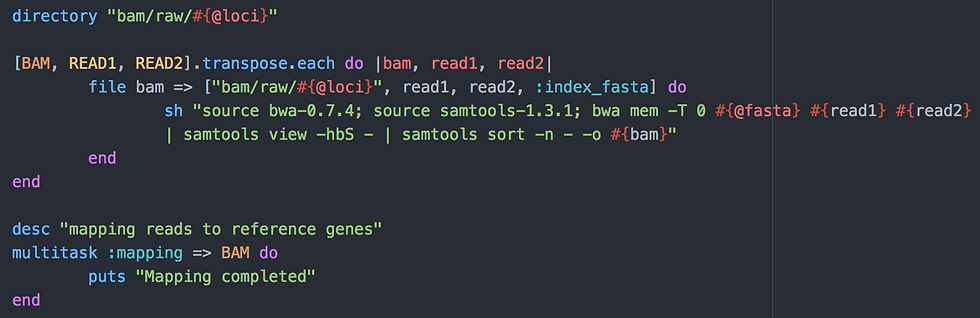
Before mapping, I need to create a folder called "bam/raw/domestication" in which the alignment (BAM) files will be stored. This is done with directory "bam/raw/#{@loci}". Then as before, I use a file task to create the BAM files. However, as I need to create many BAM files, I need to use the following little trick:
[BAM, READ1, READ2].transpose.each do |bam, read1, read2| will take the first value of the three arrays BAM, READ1 and READ2 and call them bam, read1 and read2 for use in the file task.
Imagine your first pair of FASTQ files is called "read1_1.fastq.gz" and "read1_2.fastq.gz":
the output BAM file: "bam/raw/domestication/read1"
the required input: the folder "bam/raw/domestication", my read files "read1_1.fastq.gz" and "read1_2.fastq.gz" as well as my previous task "index_fasta".
If all is here, that is the folder, the read files, the output and input files from the "index_fasta" task, the script goes on to the bwa and samtools commands. This step is pretty simple: I use bwa mem to map each pair of FASTQ files to the reference FASTA file. "-T 0" is here to keep all found alignments. That is, if a read can be mapped at, say, two different places on the reference, both are kept. Because this is not biologically possible, I will only keep the best alignment in the next filtering step. BWA outputs a SAM file which I convert to BAM using SAMtools and which I sort bfore continuing to the next step.
Again, I trigger all this by creating a task called ":mapping" which requires all my bam files (array BAM required). I also use multitask instead of task to allow my BAM files to be built in parallel.
Task 3: Filtering BAM files
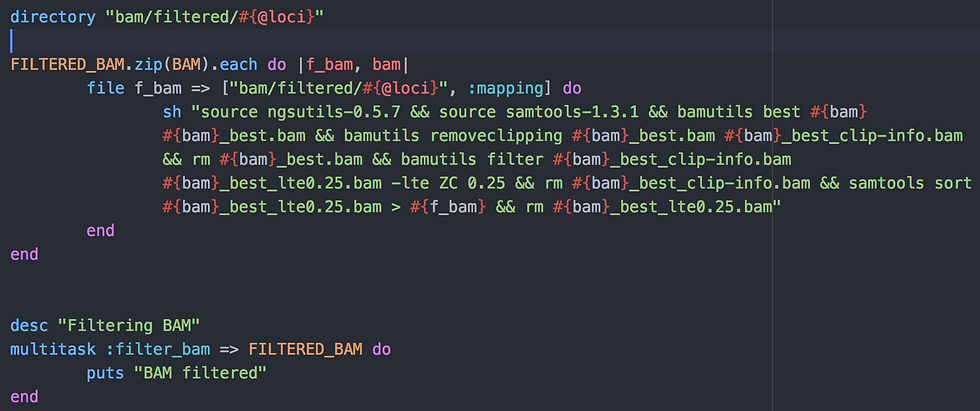
Yet another folder needs to be created where my filtered BAM files will be stored: "bam/filtered/domestication".
Then I use a method similar to transpose described above but which works well for two arrays: because FILTERED_BAM and BAM are linked in the way they are named (see Beginning of the script > Output files), you only need to link them as pairs with "zip". I name each element of the pair f_bam and bam for use in this new file task. The file task requires the folder "bam/filtered/domestication" and all input/output files from the task :mapping (which, itself, required all elements from the previous task, and so on).
The filtering steps are performed using the bamutils command of ngsutils version 0.5.7:
Keeping the best alignment when multiple were possible in previous step (lowest NM - edit distance - and best MAPQ score).
Removing reads that had more than 25% of their bases clipped by BWA. It seems that bwa mem allows the mapping of reads even if only a small portion matches the reference. This may be beneficial in some situations but I found it surprising and the step removes quite a lot of reads.
This task is called :filter_bam and requires all filtered BAMs from the array FILTERED_BAM.
End of the script
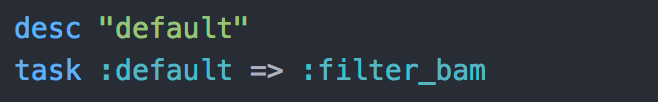
To end the script, you only need to add a default task which is the task rake will call if no task is provided from within the rake command (Beginning of the script > Usage).
In this case, the default task is the last task defined in the script. Rake will check if everything necessary for the task completion is ready. If it is, it will perform the task. If not, it will go up, one task after the other until a task can be completed. If it is the first time you run the script, Rake will go up to the beginning of the script. If it is well written, all tasks will then be completed. If not, Rake will complain and you will be able to debug. Traces are usually pretty self-explanatory so you should be fine.
Next, I will share my script to call variants using the alignment files.


Comments