My Ruby pipelines: Phasing variants (3)
- Agathe
- Jan 26, 2018
- 3 min read
Updated: Feb 9, 2019
Post 3 of the "My Ruby pipelines" series. This one is a pretty big chunk. Stay with me and you might learn a trick or two!

With the following script, you'll be able to phase variants from a VCF file. This means that, not only you'll have information on the genotypes at a particular base position but you'll also know which chromosome or haplotype each genotype call comes from. Of course, I am talking about diploid organisms here with biallelic calls.
Phasing variants can be helpful if you want to measure genetic diversity within and between populations. It adds a layer of complexity to your data, making it possible to discriminate between hypotheses. You may test for signatures of selection, estimate the incidence of recombination or hybridization between individuals or paralogs within an individual, or use software that do not allow VCF files or consider ambiguous positions in consensus sequences.
In this post, we'll split a multisample VCF file into multiple VCF files: one file per locus or chromosome, before running SHAPEIT2 to phase variants. This is done using 4 scripts that are interconnected (one command to start it all). Next, we'll reconstruct haplotypes and we'll be ready to generate population genetics statistics for each locus.
(Artwork: Aboriginal Art by Adam Reid)
Software used
VCFtools-0.1.14
shapeit-2.20
SAMtools-1.3.1
Linux commands
awk
sed
Ruby and Rake syntax
chomp & chomp!
each
ENV[]
file
FileList
File.open
flatten
join
map
multitask
pathmap
puts
readlines
Main aims
Multisample VCF file preparation
VCF file indexing
Removing non biallelic SNPs
Per locus VCF files preparation
Splitting multisample VCF file per locus
Making a list of loci with at least one SNOP
Sublisting loci with at least 10 individuals
Keeping VCF files with at least 10 individuals & one SNP
Running SHAPEIT2
Extracting Phase Informative Reads (PIRs)
Assembling (=Phasing)
Conversion to VCF
Link to script: 3b_phased_vcf-{1-4}.rake
Beginning of the script 1: 3b_phased_vcf-1.rake
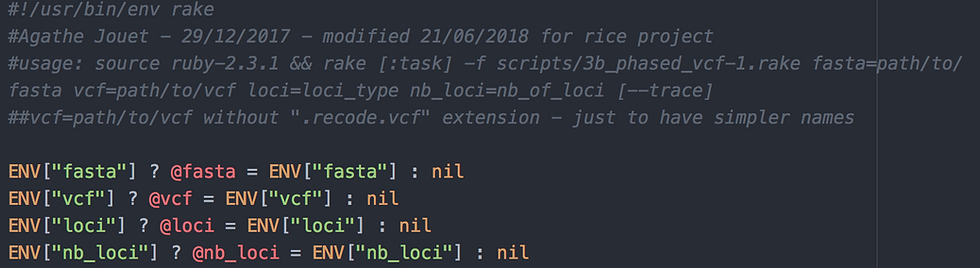
4 command line arguments are passed to ruby rake, using ENV[]:
fasta is the reference file used to create the BAM and VCF files (path required)
vcf is the name of the VCF files from which you want the variants phased (no ".recode.vcf" extension; path required)
loci is the type of loci analysed (still using the "domestication" example from 1_mapping_to_targets.rake & 2_variant_call.rake)
nb_loci is the number of loci to analyse - this will be used when calling the next rake script, to allow analysis of loci in parallel.
Task 1-1
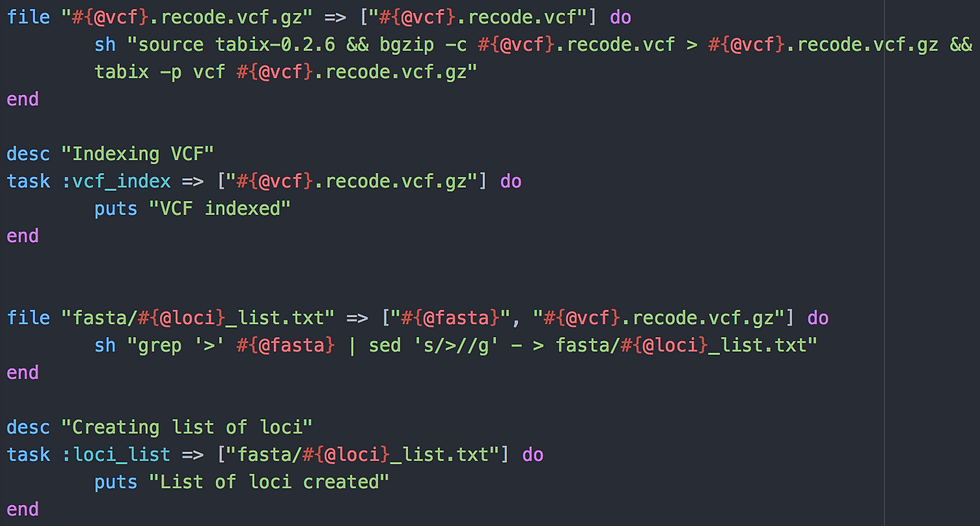
Two tasks here.
:vcf_index is used to index the multisample VCF using tabix version 0.2.6 (cannot find the doc for this particular version but v1.0 seems close enough). This is necessary if you want to use certain tools within the script
:loci_list creates the list of loci to analyse. It was done previously in 2_variant_call.rake but in case you want to start with your own bam files (not generated with the script 2), I put it there again. Could actually have been just there and removed from 2_variant_call.rake altogether. Definite room for improvement in those scripts!
Task 1-2

Two other interconnected task using VCFtools.
:remove_non_biallelic will, as the name states, remove any non-biallelic calls from the VCF file. Of course, if you are working with diploid individuals, you don't want those. An important note is that when you remove sites (any, non-biallelic or sites that are missing in too many individuals), this means that those sites will be considered as reference. You will therefore miss potential variations in your population but I find it better than to overestimate variation or to not compare like with like.
:remove_missing_snps to remove SNPs that were not biallelic in all individuals called; meaning these SNPs were gone missing now that we deleted them in the previous task.
End of script 1
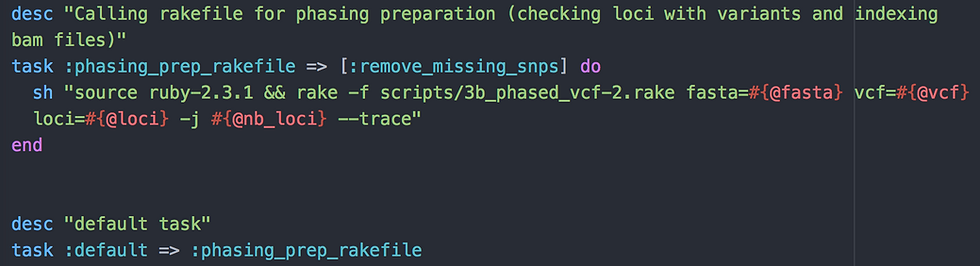
Now that we've reached the end of script 1 (3b_phased_vcf-1.rake), we need to be calling script 2 (3b_phased_vcf-2.rake). That is what we do with the default task :phasing_prep_rakefile. As for script 1, we need our reference fasta, our vcf (no extension, remember) and the type of loci. We also use the "nb_of_loci" argument you've entered for script 1 to analyse loci in parallel in the next script with the "j" flag.


Comments